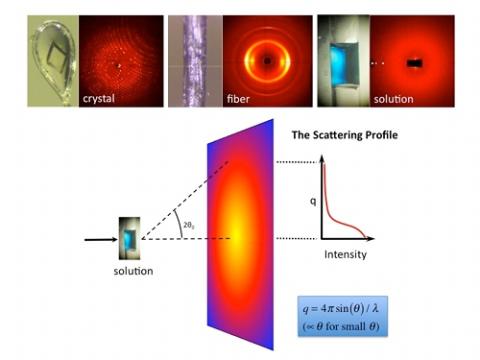
Most X-rays travel right through matter without changing direction, but approximately one out of every million gets deflected at a small angle! The very faint “small angle" scattering pattern on an X-ray detector resembles a sunset, with the undeflected beam being like the sun; there is a bright glow nearest the beam that fades to dark at higher angles. To protect our sensitive detector, we block the direct beam with a small metal beamstop. With modern algorithms, the simple pattern of light to dark on the X-ray detector can yield a surprising amount of valuable structural information about biomolecules. In fact, BioSAXS is becoming an indispensable tool in biomedical science. Not only can researchers determine the mass and size of a biomolecule in solution, but they can reconstruct its basic shape, tell if it is rigid or flexible, and even figure out how multiple molecules fit together to form complex molecular machines. Increasingly, advances in medical research depend upon gaining a clear understanding of how biomolecules function and interact within the living cell. BioSAXS is one of the few techniques that can yield structural information on how biomolecules behave under native conditions akin to that of the living cell.
What is BioSAXS?