There are four known families of membrane-immersed proteases (enzymes which break protein chains); all four carry out important functions and damage to them is implicated in pathologies including cancer, Parkinson's disease, impaired resistance to parasites, and more. When a mutation results in overactivity of a membrane protease, an inhibitor of the protease can be effective treatment for a disease. Designing such inhibitors has proven difficult, largely because of incomplete understanding of the catalytic process in the intramembrane environment.
The Urban group (Johns Hopkins) is studying E. coli GlpG, which is a member of the membrane-embedded rhomboid protease family, using measurement of enzyme kinetics coupled with crystallographic structure determinations. In previous experiments, the group found that the overall rate of the reaction (clipping off a transmembrane helix in order to release a signaling protein) is determined by the time needed for the substrate to get into the active site, rather than the reaction rate in the site ([1], see CHESS eNews #15). More recently, the focus has shifted to examining the interaction of inhibitors with GlpG. A number of complexes of rhomboid proteases with inhibitors have been examined previously, but these have all been inhibitors that bind irreversibly to the enzyme's active site and distort it in the process. The Urban group's approach is to use short peptides with an aldehyde moiety at the C-terminus; such an inhibitor binds in the active site and forms an intermediate just like the natural substrate, but is then trapped in that state.
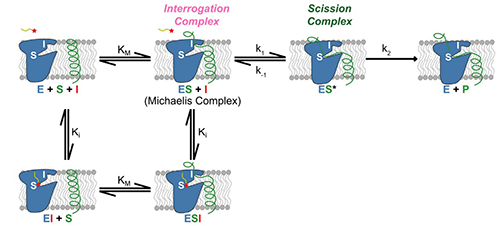
As reported in a recent publication [2], kinetic studies of the inhibition of GlpG by various peptide aldehydes showed that the inhibition is non-competitive, meaning that inhibitor and substrate can both be bound to the enzyme at once. This strongly supports the model shown in Figure 1; binding of the substrate to the “interrogation” site (which is the step that determines the reaction rate) can occur whether or not there is an inhibitor molecule bound in the “scission” site. In addition to the kinetic work, several crystal structures of protease-inhibitor complexes were determined, using data collected at CHESS. For most of these, the protease molecules were embedded in bicelles, providing an environment similar to natural membranes. A mutation (conversion of tyrosine 205 to phenylalanine) improved crystallizability with minimal effect on activity. The crystal structures revealed:
- the conformation of the tetrahedral intermediate produced when substrate (or inhibitor) binds to the catalytic serine;
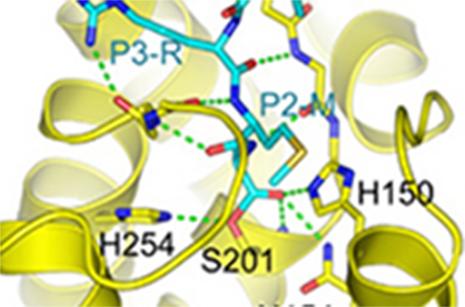
- specific interactions of the substrate/inhibitor with the protein. These largely involve backbone rather than sidechain atoms, consistent with the broad specificity of rhomboid proteases.
Several of the specific interactions involve regions outside the active site proper, as seen in Figure 2. This was unexpected, since there is no requirement for any particular residues in the P2 and P3 positions of substrates, and is helpful in designing inhibitors. The peptide aldehyde Ac-VRMA-CHO used in the structure study is in fact a potent inhibitor of GlpG in vivo, better than irreversible inhibitors used previously.
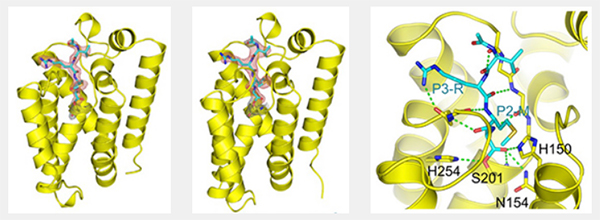
The Urban group's work suggests a strategy for improving inhibitors of rhomboid proteases: focus on reversible inhibitors such as peptide aldehydes and systematically investigate modifications to residues previously considered “nonesssential”, i.e. those which do not contact the active site.
References:
[1] S.W. Dickey, R.P. Baker, S. Cho, S. Urban, “Proteolysis inside the membrane is a rate-governed reaction not driven by substrate affinity”, Cell 155, 1270-1281 (2013).
[2] S. Cho, S.W. Dickey, S. Urban, “Crystal structures and inhibition kinetics reveal a two-stage catalytic mechanism with drug design implications for rhomboid proteolysis”, Molecular Cell 61, 329-340 (2016).