
And, the tiny beam has to hit the tiny crystal. The location of the beam can be well determined and doesn't change, but how do we know where the crystal is?
The traditional method for imaging protein crystals at most macromolecular crystallography (MX) beamlines employs an on- or off-axis digital bright-field microscope. Accurate visualization of the crystal is necessary to keep it centered in the X-ray beam as it rotates during an oscillation crystallography experiment. Precise centering becomes more difficult when the visibility of the sample is obstructed by ice or excess cryoprotectant. Several methods have been developed in the past decade to address the issues related to sample visualization on the MX beamline. Among others, raster scanning the sample with an attenuated X-ray beam and UV-excited fluorescence are growing in popularity in beamline use for the visualization of microcrystals (>10 μm). However, both UV and X-ray based methods can damage the sample during imaging due to the ionizing nature of radiation at UV and X-ray wavelengths.
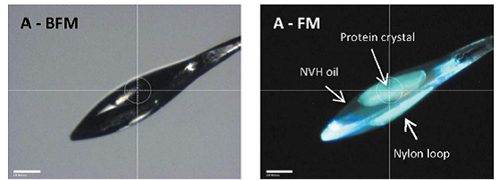
A new method for imaging protein crystals was discovered at MacCHESS and is now implemented on both the A1 and F1 stations here (Figure 1). The method utilizes visible light (405 nm) to excite conjugated double bond and delocalized electron systems natively present within a protein crystal. The magnitude of fluorescence is heavily temperature dependent with about a 10-fold stronger fluorescence at cryogenic temperatures. This novel imaging method can also be used for screening crystallization trays using fluorescence confocal microscopy (Figure 2). Unlike methods relying on non-linear optics, visible light excited fluorescence does not occur with salt crystals and thus provides a mild method for discriminating between salt and protein crystals in screening trays [1].

Reference:
[1] Lukk T, Gillilan RE, Szebenyi DME and Zipfel WR. “A visible-light-excited fluorescence method for imaging protein crystals without added dyes” J. Appl. Cryst. (2016). 49, 234–240